A variety of molecular tests are currently in use to detect oncogenic driver mutations in patients with non–small cell lung cancer (NSCLC), particularly in those with advanced-stage adenocarcinoma.1-4 For some time, molecular testing in the United States has been complicated by the regulatory environment, which is currently divided between the FDA and the Centers for Medicare & Medicaid Services (CMS).5 Tests regulated by the FDA and CMS are often used for the same purpose and in patients with the same condition, which has raised concerns that the different regulatory standards of each agency may introduce an unknown degree of variability into clinical practice.6
The FDA has historically regulated molecular tests manufactured and sold as kits by diagnostics companies, whereas CMS has overseen tests made and used within a single laboratory, called laboratory-developed tests (LDTs).5 In oncology, tests regulated by the FDA are typically called “companion diagnostics” owing to the agency’s practice of approving targeted therapies and diagnostics concurrently. The FDA approval process is designed to ensure that individual tests are accurate, reliable, and clinically valid, whereas CMS regulation under the Clinical Laboratory Improvement Amendments (CLIA) is designed to assure that tests are properly performed, largely through the oversight of laboratory personnel and procedures. Although all tests are under its jurisdiction, as a matter of policy the FDA has not actively regulated LDTs since the start of the medical device program in 1976. At the present time, companion diagnostics undergo rigorous premarket review by the FDA, whereas LDTs generally do not.
In October 2014, the FDA announced its intention to extend oversight of diagnostics to include LDTs due to the increasing complexity of LDTs and their growing role in guiding treatment decisions.7 In a Federal Register notice, the FDA stated that over 11,000 LDTs are currently used in practice.8 Yet, to date, it remains unclear how frequently LDTs are used compared with available FDA-approved tests to guide the use of targeted therapies.
We attempt to estimate the extent to which LDTs are used in NSCLC patients with advanced-stage adenocarcinoma, a setting in which molecular testing for 2 specific alterations is considered standard of care and recommended by major clinical guidelines.9,10 Testing for ALK gene rearrangements and epidermal growth factor receptor (EGFR) mutations is recommended so that patients with these genetic abnormalities can receive effective treatment with targeted agents.
Material and Methods
Study Sample Design
A universe sample frame of NSCLC-treating oncologists was created by sourcing Symphony Health Analytics’ 2014 insurance claims activity for all oncologists in the United States for both the 162 series of lung cancer ICD-9 codes as well as the claims activity related to prescribing lung cancer–targeted therapies (erlotinib, afatinib, crizotinib, and ceritinib). By combining both sources, we identified 10,184 oncologists with activity related to the care of lung cancer patients. To ensure that the physicians targeted for this research would have the required minimum number of patients to participate, we further limited this sample to those with at least 3 unique lung cancer patients in all of 2014. This reduced the list of oncologists to 8129, thus serving as the sample frame for this survey. All 8129 NSCLC-treating oncologists were invited to participate in the survey by e-mail or postal mail. Oncologists were eligible to participate if they personally managed at least 5 NSCLC patients per month and diagnosed at least 1 NSCLC patient in the past 12 months. A total of 221 oncologists responded to the survey and 153 met eligibility criteria and completed the survey. Participants were offered an industry standard honorarium as compensation for their time in completing the survey. The survey was administered online and was fielded from April 8, 2015, to September 14, 2015.
Data Collection
A questionnaire was developed to collect anonymized information on patients with stage IV NSCLC in the United States. We developed and pretested this instrument through interviews and consultations with 13 NSCLC-treating oncologists before launching the survey online. In the online survey, physicians were asked to randomly select between 3 and 8 stage IV NSCLC patients from their list of patient charts. To ensure random chart selection, oncologists were asked to choose patients whose last names began with a random selection of letters. Patient charts were required to have been active in the practice within the past 12 months to be eligible for inclusion in the study. The anonymized information collected for each patient chart consisted of the following: background information (age, weight, gender, ethnic origin, concomitant conditions, insurance type, smoking status), the year NSCLC was diagnosed, information about the genetic test (which test was used, when and in which setting was it performed, and what was the outcome of the test), and type of treatment patients subsequently received. The 153 oncologists who participated in the survey provided information for 765 patients in total. All patient chart audit data collection fields were Health Insurance Portability and Accountability Act compliant and contained no patient identifying information.
Data Analysis
All survey data were analyzed in aggregate, and the individual identities of the survey respondents were blinded to the study authors. Data were analyzed in total and split per histologic subtypes. Other dimensions such as the type of setting, geographic region, patients’ ethnic origin, insurance types, and smoking status were used to segment the analysis. The key element in the analysis was to determine, for each patient, whether a molecular test was used to identify EGFR and/or ALK alterations, and if so, whether the tests used were LDT or FDA approved. To that end, approval status of tests was determined from FDA’s publicly available list of approved companion diagnostics at the time of the survey. At the time of the survey there was no FDA-approved ROS1 test for NSCLC. Therefore, all ROS1 tests performed were qualified as LDT. Furthermore, in many instances, oncologists surveyed did not know what type of test was performed. In instances where the information was not provided by the oncologists, we followed up with the pathology lab of the relevant treating center and obtained the information by phone. We followed up with pathology labs from 96 centers and clarified the type of test for 340 of 659 EGFR-tested patients and for 288 of 562 ALK-tested patients. Data presented in this paper include the information obtained through the phone follow-up.
Ethics, Consent, and Permissions
Data for this work were obtained through market research, and no experiment on humans has been carried out. As such, there was no institutional and/or licensing committee involved in approving the experiments, and no need for informed consent from the participants, as stated in national regulations (HHS.gov; US Department of Health & Human Services; www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.html#46.102). This survey was conducted in accordance with market research guidelines such as the ones edited by the Council of American Survey Research Organizations.
Statistical Analysis
Clustered logistic regression was performed to assess whether respondent characteristics (practice setting, practice ownership, geographic region) correlated with use of an FDA-approved test. Clustering of patient records was done according to each oncologist’s group of patients.
Limitations
It should be noted that this survey has a number of limitations. First, this survey focused on oncologists, and not pathologists. The purpose of the research was to evaluate the frequency and type of testing performed and identify whether any differences in testing status were associated with patient characteristics such as age, weight, gender, ethnic origin, concomitant conditions, insurance type, smoking status, etc. We believe that oncologists are best suited to access this type of information. Topics relating to reasons for not testing a patient, number of alterations assessed (single genetic test vs next-generation sequencing), or reasons for using one type of test versus another, may largely fall with the pathologist and were outside the scope of the research. Second, our study was not designed to address the comparative outcomes of patients tested with LDTs versus FDA-approved tests. Third, while we assume that participation in the survey was random and represented basic interest and knowledge in this disease area, the potential for bias in the set of responders versus nonresponders does exist. Due to the methodology, a true response rate cannot be calculated for this survey. Physicians were invited by e-mail or postal mail, and they voluntarily self-screened based on knowledge, interest, and experience level in treating this condition. They had the opportunity to respond to the survey invitation by logging on to the online survey. As it is unknown how many physicians successfully received, reviewed, and self-screened for this survey invitation, the true response rate is unknown. Fourth, as with any survey, our findings may be influenced by response bias of the survey respondents. Although clear guidance was provided to ensure randomization of patient selection, it cannot be ruled out that some respondents might have focused on their most recent patients, or those who have been tested. Despite the potential for bias, we believe the data presented here are valuable as they represent real-life data and are usually not obtainable on a large scale. Additionally, a portion of patient records (and associated pathology reports) did not include information on the type of test used to detect lung cancer mutations and had to be excluded from further analysis (72 [14.5%] of patients tested for EGFR; 79 [16.5%] of patients tested for ALK). And last, KRAS testing rates, which predict resistance to EGFR-targeted therapy, were not evaluated because the study design predated inclusion of KRAS testing practice recommendations and guidelines.
Results
Participants
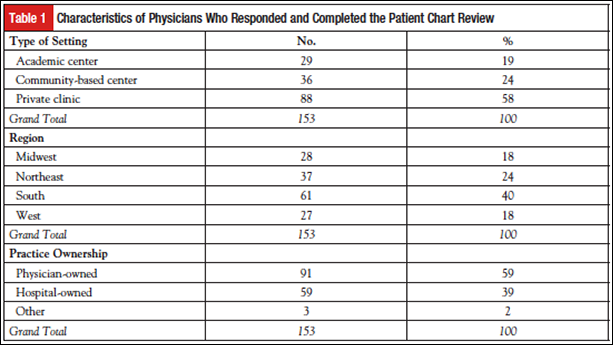
The sample of responding physicians was split across practice setting (19% academic, 24% community, 58% private) as well as geographic region and practice ownership (Table 1).
Patient Characteristics
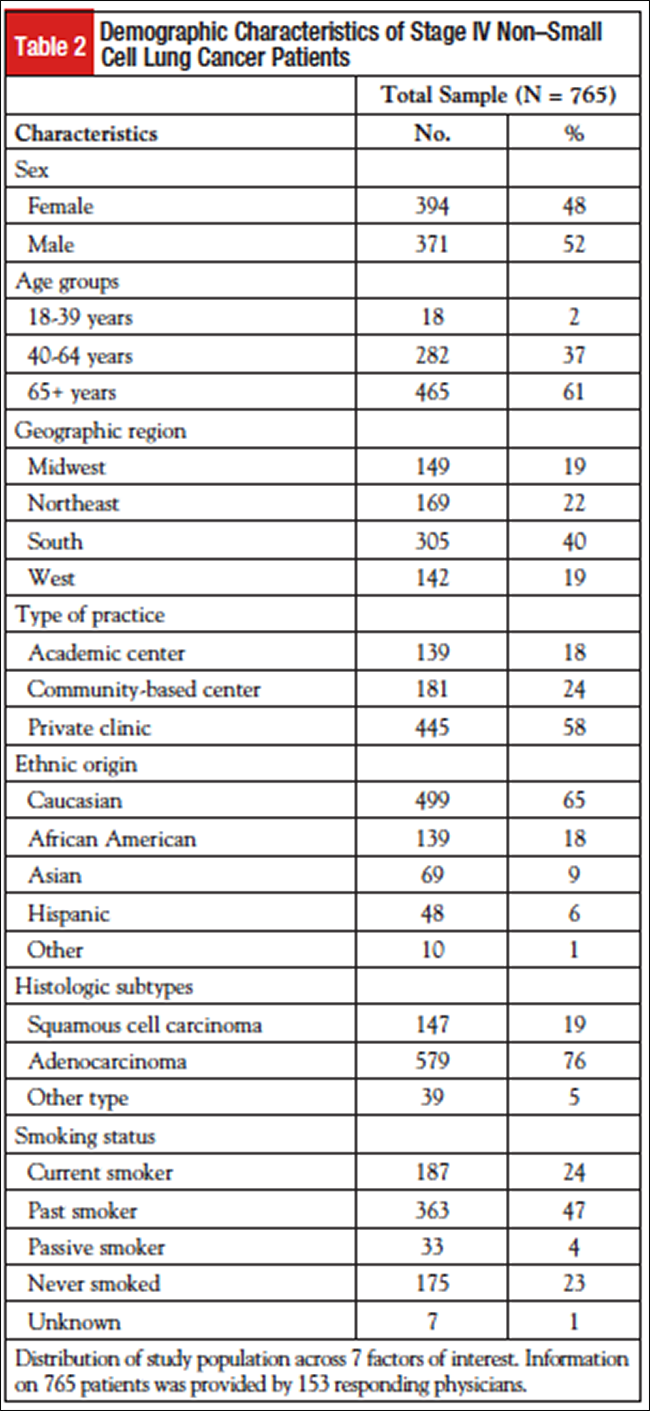
A total of 765 patients with stage IV NSCLC were reviewed in this study. The demographic characteristics of this group are presented in Table 2. Histologic subtype split was as follows: 579 (76%) of patients had adenocarcinoma, 147 (19%) had squamous cell carcinoma, and 39 (5%) had other type (including large cell and NSCLC not otherwise specified). Distribution by practice setting was as follows: 445 (58%) of patients were followed in privately owned clinics, 181 (24%) in community-based centers, and 139 (18%) in academic medical centers. Fifty-two percent of patients were male, and 61% were aged 65 years or older.
Overall Test Rate
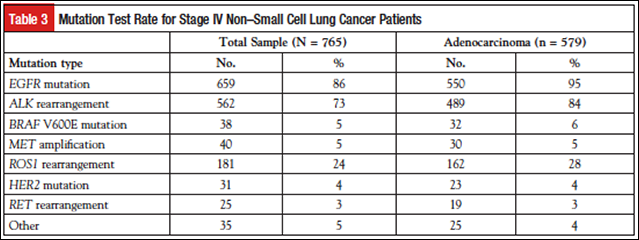
Among the 579 patients with adenocarcinoma, 550 (95%) and 489 (84%) were tested for EGFR mutations and ALK rearrangements, respectively (Table 3). Other genetic alterations (BRAF, MET, HER2, RET) were tested at lower frequencies, with one exception being ROS1 gene fusion testing at 28% of adenocarcinoma patients.
Use of FDA-Approved Tests
Of the 550 adenocarcinoma patients tested for EGFR mutations, 496 (90%) were diagnosed or tested following the first FDA approval of an EGFR test for lung cancer on May 14, 2013. Seventy-two patients had an unknown test type and were excluded from further analysis. Of the remaining 424 patients, 55 (13%) received an FDA-approved test and 369 (87%) received an LDT (Table 4). We performed a similar analysis for adenocarcinoma patients tested for ALK rearrangements. Of the 489 adenocarcinoma patients tested for ALK, 478 (98%) were diagnosed or tested on or after August 26, 2011, the date of the first drug-diagnostic approval for NSCLC with detected ALK rearrangement. Excluding 79 patients with unknown test type, 204 (51%) patients received an FDA-approved test while 195 (49%) were tested with an LDT (Table 4).

Statistical Analysis
The characteristics practice setting, practice ownership, and geographic region were evaluated for correlation with use of an FDA-approved test. None of the characteristics reached nominal statistical significance (P <.05) for use of either an FDA-approved EGFR or ALK test.
Discussion
This study was undertaken to evaluate the prevalence of molecular testing in lung cancer, as well as the use patterns of tests overseen by different regulatory agencies. Although much has been written about the rate of molecular testing in oncology and in lung cancer specifically, little is currently known about the relative use of FDA- versus CLIA-regulated tests (the latter are referred to as LDTs in this article). This study seeks to address that gap by viewing lung cancer as a case study, owing to the diversity of testing options that exist in that setting. Findings from this study will help inform the debate over how best to structure regulatory oversight of molecular testing in the future.
The patient chart review conducted in this study revealed that a large proportion of patients with advanced lung adenocarcinoma underwent molecular testing for EGFR mutations and ALK rearrangements in accordance with major clinical guidelines.9,10 The high rate of overall testing observed in this study is consistent with other findings in the literature11-13 and supports the claim that molecular testing is now a routine part of advanced lung cancer treatment. Moreover, the finding that ROS1 was the third most commonly tested biomarker is not surprising given the fact that ROS1 has been recognized as a potential therapeutic target for some time14 and was approved as a target for crizotinib in March of 2016.15
This study also found that testing was more commonly performed with LDTs than with FDA-regulated tests for EGFR mutations and was evenly split between LDTs and FDA-regulated tests for ALK rearrangements. The high rate of LDT use may be caused by a number of factors. First, clinical guidelines are not prescriptive about specific testing platforms. It remains unknown whether there is any quality trade-off associated with the use of many commonly used LDTs in place of FDA-regulated tests in settings where both exist, and both FDA-regulated tests and LDTs are generally considered acceptable so long as proven test methodologies are used.16 Second, many LDTs became available prior to the introduction of FDA-approved alternatives. This was the case, for example, with tests for EGFR mutations in lung cancer, where the first EGFR-targeted therapy was approved several years prior to FDA clearance of an EGFR test, leading to the introduction of LDTs17 for EGFR prior to the approval of the cobas EGFR Mutation Test. As a result, physicians may have developed comfort and familiarity with the LDT prior to the availability of the FDA-approved test. Third, many tumor biopsies provide limited tissue for testing, which may encourage the use of assays that detect multiple biomarkers simultaneously, none of which are currently FDA approved for use in lung cancer. This study did not collect information on the cost of tests, and we cannot speculate on whether cost plays a role in the decision to use an FDA-regulated test or an LDT.
There are pros and cons to the widespread use of LDTs. On the one hand, LDTs may offer rapid technological advances and facilitate innovation in molecular testing and have been demonstrated in some cases to offer advantages beyond existing FDA-regulated alternatives.18,19 On the other hand, concerns exist that LDTs are not currently subjected to premarket review by the FDA and thus are not required to meet the same evidentiary standards as FDA-regulated tests. Additionally, LDTs have in at least some instances been reported to perform poorly, as noted in a report of case studies released by the FDA.20 Our study, however, does not seek to address the relative quality of LDTs and FDA-regulated tests, but rather the relative frequency of use.
Owing to the large number of tests currently in use, some of which have been subjected to premarket review by the FDA while others have not, there exists the potential for wide variability in test performance and claims.21-23 As demonstrated by this study, LDTs and FDA-regulated tests are often used in the same setting, raising the concern that an unknown degree of variability may exist between tests for the same intended use.
Steps should be taken to mitigate uncharacterized variability between tests used in the same clinical setting. Further evaluation of the relative performance of tests intended to measure the same alteration is needed to identify cases in which different tests may not provide comparable results.
Acknowledgments
We thank Hui Jiang, PhD, Professor of Biostatistics, University of Michigan, for performing the regression analysis for this study. We also thank Travis Deseran, Associate Director, Deerfield Institute, for preparing and organizing the data file for this study.
Conflict of Interests
Authors declare no conflicts of interest.
References
- Gray S, Hicks-Courant K, Cronin A, et al. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32:1317-1323.
- Pao W, Kris MG, Iafrate AJ, et al. Integration of molecular profiling into the lung cancer clinic. Clin Cancer Res. 2009;15:5317-5322.
- Sequist LV, Heist RS, Shaw AT, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616-2624.
- Morgensztern D, Campo MJ, Dahlberg SE, et al. Molecularly targeted therapies in non–small-cell lung cancer annual update 2014. J Thorac Oncol. 2015;10:S1-S63.
- Weiss RL. The long and winding regulatory road for laboratory-developed tests. Am J Clin Pathol. 2012;138:20-26.
- Sharfstein J. FDA regulation of laboratory-developed diagnostic tests: protect the public, advance the science. JAMA. 2015;313:667-668.
- US Food and Drug Administration. Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs): Draft Guidance. www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/
ucm416685.pdf. Accessed March 31, 2016. - US Food and Drug Administration. Food and Drug Administration Notification and Medical Device Reporting for Laboratory Developed Tests; Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories; Availability. www.regulations.gov/#!documentDetail;D=FDA-2011-D-0357-0001. Accessed March 31, 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Non-Small Cell Lung Cancer. Version 4.2016. www.nccn.org/store/login/login.aspx?ReturnURL=http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed March 31, 2016.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn. 2013;15:415-453.
- Lim C, Tsao MS, Le LW, et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall cell lung cancer. Ann Oncol. 2015;26:1415-1421.
- Volmar KE, Idowu MO, Souers RJ, et al. Molecular testing in anatomic pathology and adherence to guidelines: a College of American Pathologists Q-Probes study of 2230 testing events reported by 26 institutions. Arch Pathol Lab Med. 2015;139:1115-1124.
- Schink JC, Trosman JR, Weldon CB, et al. Biomarker testing for breast, lung, and gastroesophageal cancers at NCI designated cancer centers. J Natl Cancer Inst. 2014;106:dju256.
- Politi K, Herbst RS. Lung cancer in the era of precision medicine. Clin Cancer Res. 2015;21:2213-2220.
- US Food and Drug Administration. FDA expands use of Xalkori to treat rare form of advanced non-small cell lung cancer. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm490329.htm. March 11, 2016.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Guideline. J Clin Oncol. 2014;32:3673-3679.
- Benlloch S, Botero ML, Beltran-Alamillo J, et al. Clinical validation of a PCR assay for the detection of EGFR mutations in non-small-cell lung cancer: retrospective testing of specimens from the EURTAC trial. PLoS One. 2014;9:e89518.
- Association for Molecular Pathology. Facts FDA Ignored: An analysis of the FDA report, “The Public Health Evidence for FDA Oversight of Laboratory Developed Tests: 20 Case Studies.” http://amp.org/emailads/documents/AMPResponseFDACaseReportFinal.pdf. 2015. Accessed May 8, 2016.
- Evans JP, Watson MS. Genetic testing and FDA regulation: overregulation threatens the emergence of genomic medicine. JAMA. 2015;313:669-670.
- US Food and Drug Administration. The Public Health Evidence for FDA Oversight of Laboratory Developed Tests: 20 Case Studies. www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UCM472777.pdf. November 11, 2015.
- Conway P. Congressional Testimony before the Committee on Energy and Commerce, Subcommittee on Health, US House of Representatives. Examining the regulation of diagnostic tests and laboratory operations. November 17, 2015.
- Yorczyk A, Robinson LS, Ross TS. Use of panel tests in place of single gene tests in the cancer genetics clinic. Clin Genet. 2015;88:278-282.
- Polley MY, Leung SC, McShane LM, et al. An international Ki67 reproducibility study. J Natl Cancer Inst. 2013;105:1897-1906.