 Myelodysplastic syndromes (MDS) define a group of hematologic disorders whose pathologic findings have been well defined.1 Controversy exists, however, in the reproducibility of the various subtypes. The 4th edition of the World Health Organization’s WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues was last published in 2008.2,3 The classification included morphology, clinical parameters, immunophenotyping, and genetics. The 2016 revision (5th edition) of the WHO classification of myeloid neoplasms and acute leukemia was published in May 2016, and the changes will be discussed in this article.4
Myelodysplastic syndromes (MDS) define a group of hematologic disorders whose pathologic findings have been well defined.1 Controversy exists, however, in the reproducibility of the various subtypes. The 4th edition of the World Health Organization’s WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues was last published in 2008.2,3 The classification included morphology, clinical parameters, immunophenotyping, and genetics. The 2016 revision (5th edition) of the WHO classification of myeloid neoplasms and acute leukemia was published in May 2016, and the changes will be discussed in this article.4
The foundation for the diagnosis of MDS relies on the morphological dysplasia involving 1 or more cell lines and ineffective clonal hematopoiesis that result in peripheral blood cytopenias. MDS are inherently volatile and can transform to acute myeloid leukemia (AML). In current terminology, MDS patients are defined by cytopenia(s) as defined in the original International Prognostic Scoring System (IPSS; hemoglobin <10 mg/dL, absolute neutrophil count [ANC] <1.8 x109/L, or platelets <100 x 109/L). In addition, there must be less than 20% myeloblasts in the bone marrow, ≥10% dysplasia in 1 or more of the lineages, evidence of clonality and/or an abnormal karyotype typical for MDS.5 Even among expert pathologists, there has been interobserver variation, and there can be difficulty in distinguishing MDS from non-neoplastic disorders.6 One must exclude vitamin B12 and folate deficiency, HIV or other infection, alcohol abuse, medications such as methotrexate and chemotherapy, copper deficiency, autoimmune disorders such as idiopathic thrombocytopenic purpura, large granular lymphocyte disorders, Fanconi’s anemia, and even aplastic anemia and myeloproliferative disorders.7 The clonal dysplastic features of the hematopoietic cell lines are essential to the WHO definition of MDS. However, except for MDS associated with isolated del(5q), the diagnosis of MDS has not yet been established by chromosomal abnormalities or discrete gene abnormalities because most of the chromosomal abnormalities and/or discrete genetic lesions are not specific to a diagnosis of MDS (ie, can be seen in AML and even in nonmyeloid malignancies), and therefore the use of these modalities to confirm a diagnosis of MDS is unreliable.8
MDS are classified in the bone marrow by first obtaining a well-stained bone marrow aspirate. According to the WHO criteria, 500 cells should be counted. Although not specific to MDS, the current classification paradigm includes ≥10% of a hematopoietic cell line as dysplastic. Common findings in the morphology include neutrophil hypogranularity, erythroid abnormalities that include an altered nuclear:cytoplasmic ratio, nuclear budding, bilobed nuclei, cytoplasmic vacuoles, and small hypolobulated megakaryocytes.9 Goasguen et al have proposed changes for refining the definition of dysgranulopoiesis and dysmegakaryopoiesis in the MDS.10,11
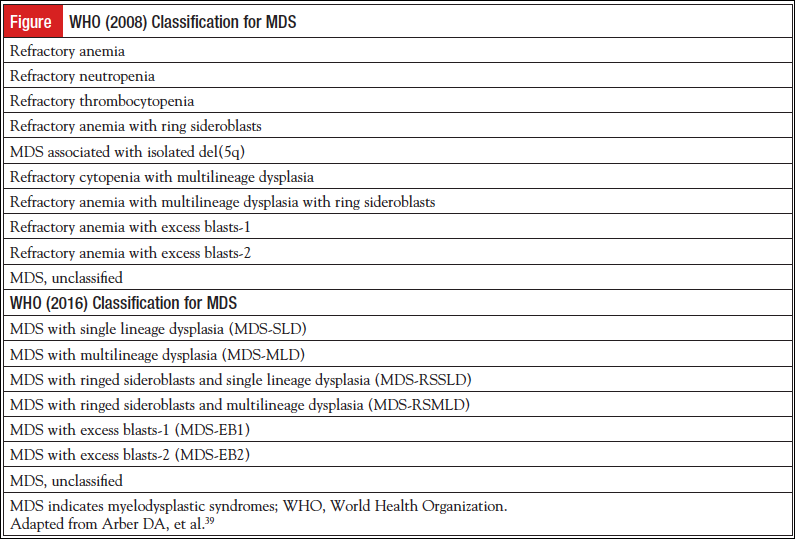
The morphological changes previously described in MDS remain the same in the WHO (2016) revision; however, the nomenclature did change. The terms refractory anemia and refractory cytopenia will be replaced by MDS.12 Currently MDS with isolated del(5q) is the only MDS entity defined by a cytogenetic abnormality.13
Conventional cytogenetics are included as an obligatory prerequisite for the evaluation and prognosis of suspected MDS.14-16 Using conventional cytogenetic techniques, about 50% of de novo MDS patients will be found to have a chromosomal abnormality. There is an inverse correlation between the number of chromosomal abnormalities and the median survival. By expanding the database, additional cytogenetic abnormalities were incorporated into the revised IPSS (IPSS-R). Schanz et al developed a cytogenetic classification scoring system based on 2902 untreated, de novo MDS patients.17 The cytogenetic abnormalities will remain the same as listed in WHO (2008). There is, however, a revision in the MDS characterized by del(5q). This entity is known to have a favorable prognosis and a high response rate to lenalidomide.18 One additional chromosomal abnormality (except monosomy 7) does not affect survival; however, ≥2 chromosomal abnormalities will markedly reduce survival from 58 months to 6.8 months.19
Fluorescence in situ hybridization (FISH) is a cytogenetic technique that uses fluorescent probes that only bind to those parts of the chromosome with a high degree of complementarity. It is used to detect and localize the presence or absence of specific chromosomal translocations and gains and losses of DNA segments that are commonly found in MDS. FISH can be used on nondividing cells, and can even be used in identifying and risk-stratifying chromosomal abnormalities in the peripheral blood when a bone marrow is not obtainable. FISH does not appear to add a significant amount of information to conventional cytogenetics.20 The relevance of small clones detected only by FISH and not karyotyping is not established. Flow cytometry immunophenotyping will not be required in the WHO (2016) revision; however, it may provide useful information in difficult cases. A CD34+ count is relevant in high-risk disease. If the information is relevant, it should become part of the final report.21
MDS with ringed sideroblasts with single lineage dysplasia (MDS-RSSLD) or multilineage dysplasia (MDS-RSMLD) represents a significant change in the WHO (2016) revision.10 The percentage of ring sideroblasts does not correlate with prognosis.22 In the WHO (2008) classification, there was refractory anemia with ring sideroblasts (RARS), and the category refractory cytopenia with multilineage dysplasia with ring sideroblasts (RCMD-RS) was incorporated into the category of refractory cytopenia with multilineage dysplasia (RCMD), since the prognosis of RCMD was similar to RCMD-RS.21 Mutations in the spliceosome of gene SF3B1 has been strongly associated with ring sideroblasts.23,24 Since RARS and RCMD-RS express the SF3B1 gene mutation, share a unique gene expression profile, and show ring sideroblasts, the WHO (2016) classification will retain the WHO (2008) category of RARS, but it is now called MDS-RSSLD if 1 cell line is dysplastic, with a new category of MDS-RSMLD if more than 1 cell line is dysplastic.10 Ring sideroblasts are not unique to MDS and can be seen in refractory anemia with excess blasts and even in AML; however, these entities are defined by their blast counts.25 The identification of the SF3B1 gene mutation, when present, will allow the diagnosis of MDS-RSSLD or MDS-RSMLD when ring sideroblasts are present, but less than 15%.4,26 The SF3B1 gene mutation will not be required for diagnosis; however, in patients found to have ≥5% ring sideroblasts and the presence of this mutation, a diagnosis of MDS-RS can still be made. If the SF3B1 mutation is not present, there still must be ≥15% ring sideroblasts.
MDS-unclassifiable (MDS-U) includes cases with 1% blasts in the peripheral blood (repeated on at least 2 occasions), <5% blasts in the bone marrow, and evidence of at least 1 dysplastic lineage and at least 1 cytopenia as defined by the IPSS criteria, and no Auer rods in the bone marrow. The other categories include single lineage dysplasia and pancytopenia by the IPSS criteria, bone marrow with <5% blasts, peripheral blood with <1% blasts, and no Auer rods. This would include isolated del(5q) and pancytopenia. Ring sideroblasts can be present in MDS-U, but they must represent <15% of the marrow erythroid elements, the SF3B1 mutation must be present, no dysplastic lineages, at least 1 cell lineage is cytopenic by the IPSS criteria, the bone marrow has <5% blasts, the peripheral blood contains <1% blasts, and there are no Auer rods.4
Erythroleukemia (erythroid/myeloid type) was defined in WHO (2008) as having at least 50% erythroid precursors in the entire marrow nucleated cell population and myeloblasts that account for at least 20% of the total nonerythroid cell population. There has to be >20% myeloblasts in the total cell population.27 This has caused confusion, because you can still have a high number of erythroid precursors and a low total bone marrow myeloblast count and still allow the diagnosis of acute leukemia.28 The erythroid/myeloid type of acute erythroid leukemia has been divided into different prognostic subsets based on their cytogenetic and molecular genetic features.29 In the WHO (2016) edition, the nonerythroid blast count criteria will be eliminated from erythroleukemia of the myeloid/erythroid type, and if the total myeloid blast count is <20%, the case can be classified into the proper MDS category. Pure erythroid leukemia will be the only type of acute erythroid leukemia and will remain a subtype of AML not otherwise specified subtype.
The natural history of MDS varies considerably between individuals, which correlates with the mosaic of subtypes of MDS.30 Some patients live months, whereas others live years. There are 3 FDA-approved agents for the treatment of MDS—5-azacytidine, decitabine, and lenalidomide. Immunosuppressive medications and allogeneic stem cell transplantation are also used. One of the greatest challenges with our present therapeutics is to balance the current therapeutic benefit of treatment with the associated toxicities of treatment. Designing models to accurately predict an individual’s survival is predominant and essential to guide the proper timing and selection of the therapeutic.
Current models for risk stratification were limited by information that could be obtained from examination of the complete blood count, the examination of the bone marrow, and conventional cytogenetics. The 1997 IPSS is still used as a standard prognostic tool in clinical trials.31 The prognosis relies on the sum of 3 components to calculate a score: the blast count, the karyotype, and the number of peripheral blood cytopenias. The score stratifies patients into risk groups, which are correlated with their overall survival. The higher the score, the shorter the survival. There are 4 assigned risk groups: lower-risk MDS included low risk and intermediate (INT)-1, and higher risk included INT-2 and high risk, based on the scoring system. This type of scoring system was used in subsequent models to estimate an individual’s expected survival. This first attempt at a classification scheme for MDS had several deficiencies that led to other models.32 Specifically, there was a limited number of karyotypes; transfusion dependence was not included; prior therapy, the degree of cytopenias, and the spectrum of dysplasia were not considered; and patients with chronic myelomonocytic leukemia/myeloproliferative neoplasm (CMML/MPN) or therapy-related MDS (t-MDS) were not included. The IPSS included a category of 20% to 30% blasts, which has now been reclassified as AML with myelodysplastic features. The model only included newly diagnosed patients with MDS. The IPSS-R was published in 2012.33 The IPSS-R included more prognostic karyotypes, such as the percentage of blasts, hemoglobin level, platelet count, and ANC. In the IPSS-R scoring system, more significance was given to the cytogenetics than in the IPSS.
While the 3 essential elements of the IPSS—the bone marrow blast percentage, cytogenetics, and cytopenias—remained, the definition and scoring of each category was refined. The bone marrow blast percentage of ≤5% was divided, certifying that even a small number of blasts in the circulation was an adverse risk factor. The cytogenetic stratification allowed for more proportional influence than in the IPSS. The degree of a cytopenia was more relevant than just an affected cell lineage. The categories were calculated into the very low, low, intermediate, high, and very high risk groups. Both the IPSS and the IPSS-R calculated the median survival time for 25% of the patients to transform to AML.
Because of the wide variation in each IPSS and IPSS-R group, other MDS prognostic scoring systems were developed to provide survival information during the entire course of the disease. The WHO classification–based Prognostic Scoring System (WPSS) 2007 included information from the WHO classification scheme, karyotypes as described in the original IPSS categories, and transfusion dependence.34 The WPSS could be used at diagnosis or at a future time point. Some of the drawbacks of this scoring system include the lack of an absolute white blood count and platelet count, the lack of the expanded cytogenetics as was seen in IPSS-R, and the absence of any criteria to support a blood transfusion.35 This scoring system has never been validated in patients with CMML, t-MDS, or MPS/MPN syndromes.
The Global MD Anderson Cancer Center MDS Prognostic Model (2008) was designed to allow the evaluation of all suspected MDS patients at any time and included t-MDS, CMML, and MDS/MPN.36 This model improved on the prognostic accuracy of the IPSS; however, the scoring system was complex, and only 2 abnormal karyotypes were included. About 20% of the low-risk patients with MDS as categorized by the IPSS scoring system had aggressive disease and a shortened survival. The MD Anderson Low-Risk Prognostic Scoring System (LR-IPSS+, 2008) was able to classify IPSS low and INT-1 risk into 3 risk groups using 5 prognostic factors.37 This led to 3 subsets of survival for a median of 80.3 months, 26.6 months, and 14.2 months. The treatment-related MDS Prognostic Scoring System (T-PSS) and the 2008 WHO subdivided patients with t-MDS into prognostic groups.2,38 Inclusion of these patients in a prognostic scoring system was important because they had been excluded from the IPSS, IPSS-R, and WPSS, and some categories appear to be inherently less responsive and have a shorter survival than untreated MDS. (For a table of the current risk stratification models, see reference 39.)
Since about 50% of the MDS patients will not have an identifiable chromosomal abnormality detected by conventional techniques, next-generation sequencing (NGS) has identified recurrent genetic mutations in up to 90% of MDS patients and will be the next leap in the identification and the risk prognostication of MDS.40 The most common genetic mutations are found in pathways that affect pre-mRNA splicing (SF3B1,U2AF1,SRSF2), tumor suppressors (TP53, MYBL2, BLU), epigenetic regulators (TET2, ASXL1, DNMT3A, EZH2), impaired differentiation (RUNX1, SETBP1, ETV6), and proliferation (JAK2, NRAS, CDKN2A, PTEN).41 Point mutations in EZH2, ETV6, ASXL1, RUNX1, and NRAS are associated with a poor survival, and somatic mutations in TP53 and DNMT3A have a very poor prognosis, especially with an allogeneic transplantation; however, SF3B1 mutations are associated with a better outcome. An increased number of mutations in a given individual with MDS is associated with shortened survival and an increased risk of developing AML.42 As stated, SF3B1 gene mutations have been seen in MDS with ring sideroblasts, but it has also been associated with thrombocytosis and with JAK2, CALR, and MPL exon 10 mutations.43,44 TP53 gene mutations have been identified in about 20% del(5q), have an increased risk of transformation to AML, are associated with a poor response to lenalidomide, and have a poor postallogeneic transplant outcome.45 TET2 gene mutations are associated with an improved response to treatment with 5-azacytidine but a poor postallogeneic transplant outcome.46 Spliceosomal mutations in SRSF2, SF3B1, and U2AF1 are associated with a poor prognosis in chronic myelomonocytic leukemia.47
Most patients with MDS will not die of AML but of complications of their cytopenias or age-related conditions that are common in the geriatric population, such as cardiovascular disease.48 Risk stratification will become even more accurate in the future with molecular genetic data. Haferlach et al developed a risk stratification model based on the mutational status of 14 genes in 944 patients with MDS combined with variables found in the IPSS that has the potential to prognosticate independently of the IPSS score.49 Another study by Bejar used a different model of clinical and genetic predictors to determine prognosis in MDS.50
The diagnosis of MDS can be difficult for a number of reasons, including: the degree of dysplasia may be minimal or even undetectable; certain myeloid and lymphoid disorders can either mimic or co-occur with MDS; over 80% of early MDS patients lack a specific chromosomal abnormality and therefore have no evidence of clonality, have a mild cytopenia without dysplasia or a cytogenetic abnormality, or a typical karyotype but only mild or no dysplasia.51 Molecular genetic panels have been developed that reflect the most common somatic mutations seen in MDS to further clarify the diagnosis of MDS.49 In the recent past, more academic and commercial labs are offering gene mutation testing from patients with cytopenias. The critical disease genes known to occur in MDS are grouped by their functional type and can aid in establishing the clonal nature of MDS. An abnormal gene mutation in a driver mutation can suggest MDS when 1 or more cytopenias are present but the classic criteria for MDS is not present, such as ≥10% dysplasia in 1 or more cell lineages, 5% to 19% blasts in the bone marrow, an abnormal karyotype typical for MDS, or evidence of clonality.
Patients who fail to meet the criteria for MDS are preliminarily classified as Idiopathic Cytopenias of Undetermined Significance (ICUS).52 This describes an entity in which MDS is suspected but not proved. This provisional category requires a significant cytopenia in 1 or more of the hematopoietic cell lines: hemoglobin <11.0 g/dL, ANC <1000/µL, or platelets <100 × 109, and none of the classic findings of MDS such as ≥10% dysplasia in any cell line, myeloblasts ≥5% of the marrow cellularity, and no acquired chromosomal abnormality specific for MDS or AML. The findings must be present for at least 6 months, the cytopenia cannot be explained by any other reason, and the bone marrow does not meet the criteria for MDS. Unlike monoclonal gammopathy of undetermined significance or monoclonal B-cell lymphocytosis, clonality is not required. Some patients with ICUS will progress to MDS, and progression to a myeloproliferative disorder has also been described. There exists a subset of patients with ICUS who do not demonstrate clonality but do not fulfill the criteria for MDS. The proposed criteria for Clonal Cytopenias of Undetermined Significance (CCUS), as described by Kwok et al, include the same criteria for the blood and bone marrow as for ICUS, however, there is 1 or more of the following genetic findings, which include an acquired chromosomal abnormality that is not diagnostic of MDS or AML: an acquired chromosomal abnormality not diagnostic of a hematologic malignancy, and the presence of a somatic mutation with a variant allele frequency (VAF) ≥2% in a hematologic malignancy–associated gene in the peripheral blood or bone marrow.53 They examined 144 patients who were sent to Genoptix Medical Laboratory with a diagnosis of unexplained cytopenias. The subjects were classified into MDS, ICUS with mild dysplasia, and ICUS with no dysplasia, and also used a 22–myeloid gene panel consisting of common genes found in myeloid malignancies. Based on conventional hematopathology, 17% were diagnosed with MDS, 15% with ICUS with some evidence of dysplasia, and 69% with ICUS with no evidence of dysplasia. The bone marrow DNA was sequenced for the 22 frequently mutated genes and identified 71% of the MDS patients, 62% of the ICUS patients with some dysplasia, and 20% of the ICUS patients with no dysplasia. Thirty-five percent of the ICUS patients demonstrated a somatic mutation or a chromosomal abnormality and were classified as CCUS. The most common somatic mutations in MDS and ICUS were SF3B1 and TET2 mutations. The findings were confirmed in a cohort of 91 lower-risk MDS and 249 ICUS cases. Somatic mutations were found in 79% of the MDS patients, 45% of the ICUS patients with dysplasia, and 17% of the ICUS patients without dysplasia. ICUS was more than 5 times more common than MDS, and CCUS was more common than MDS.
Unfortunately, there is no specific somatic mutation that will differentiate MDS from clonal hematopoiesis of indeterminate potential or CCUS except SF3B1 mutations, which are associated with ringed sideroblasts. Cargo et al attempted to distinguish characteristics that would identify those patients with early MDS from healthy individuals.54 They identified 69 individuals who had a diagnosis of ICUS and subsequently developed MDS or AML. They performed targeted sequencing and array-based cytogenetics and were able to identify a driver mutation and/or a structural variant in 91% of the ICUS patients who progressed to MDS or AML. They state that the lack of a control group limits the diagnostic utility of their findings.
Because about 90% of MDS patients carry ≥1 oncogenic mutations and two-thirds of them are found in individuals with a normal karyotype, it would be useful if the gene mutation was specific to a category of MDS or MDS-like disease. However, it has been shown that about 10% of patients 70 to 79 years of age and 20% of persons 80 years or older have clonal somatic mutations detectable at a VAF of ≥2%. This entity has been preliminarily classified as Clonal Hematopoiesis of Indeterminate Potential (CHIP).55-57 Like monoclonal gammopathy of undetermined significance and monoclonal B-cell lymphocytosis, the transformation rate of CHIP to a hematologic malignancy is about 0.5% to 1% a year. Although CHIP is associated with an increased all-cause mortality, this entity is not MDS with certainty. Some somatic mutations have been identified in patients without cytopenias. Some of the frequently mutated genes in CHIP, such as SF3B1, TP53, DNMT3A, TET2, SRSF2, and ASXL1, are also seen in myeloproliferative disorders and AML. Even the high-risk mutated genes associated with a poor prognosis, such as ASXL2, RUNX1, DNMT3A, and TP53, are seen with equal frequency in MDS, ICUS, and CCUS.
Potentially, risk stratification will become more accurate in the future with the incorporation of NGS, comorbidities, and other prognostic biomarkers. Hopefully, this will translate into better predictive models for more targeted therapeutic decisions. This represents an ideal paradigm to incorporate precision medicine into daily practice.
References
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361:1872-1885.
- Brunning RD, Orazi A, Germing U, et al. Myelodysplastic Syndromes/Neoplasm. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008:89-107.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-951.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405.
- Greenberg P, Cox C, LeBeau MM, et al. International scoring for evaluating the prognosis in myelodysplastic syndromes. Blood. 1997:89;2079-2088.
- Font P, Loscertales J, Beavente C, et al. Inter-observer variance with the diagnosis of myelodysplastic syndromes (MDS) following the 2008 WHO classification. Ann Hematol. 2013;92:19-24.
- Steensma DP. Dysplasia has a differential diagnosis: distinguishing genuine myelodysplastic syndromes (MDS) from mimics, imitators, copycats and impostors. Curr Hematol Malig Rep. 2012;7:310-320.
- Neuman WL, Rubin CM, Rios RB, et al. Chromosomal loss and selection are the most common mechanisms for loss of heterozygosity from chromosomes 5 and 7 in malignant myeloid disorders. Blood. 1992;79:1501-1510.
- Matsuda A, Germing U, Jinnai I, et al. Improvement of criteria for refractory cytopenia with multilineage dysplasia according to the WHO classification based on the prognostic significance of morphological features in patients with refractory anemia according to the FAB classification. Leukemia. 2007;21:678-686.
- Goasguen JE, Bennett JM, Bain BJ, et al. Proposal for refining the definition of dysgranulopoiesis in acute myeloid leukemia and myelodysplastic syndromes. Leuk Res. 2014;38:447-453.
- Goasguen JE, Bennett JM, Bain BJ, et al. Quality control initiative on the evaluation of dysmegakaryopoiesis in myeloid neoplasms: difficulties in the assessment of dysplasia. Leuk Res. 2016;45:75-81.
- Arber D, Hasserjian RP. Reclassifying myelodysplastic syndromes: what’s where in the new WHO and why. Hematology Am Soc Hematol Educ Program. 2015;2015:294-298.
- Germing U. Lauseker M, Hildebrandt B, et al. Survival, prognostic factors and rates of leukemic transformation in 381 untreated patients with MDS and del(5q). Leukemia. 2012:26:1286-1292.
- Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385-4395.
- Sole F, Luño E, Sanzo C, et al. Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica. 2005;90:1168-1178.
- Ohyashiki K, Kodama A, Ohyashiki JH. Cytogenetics in myelodysplastic syndromes. Methods Mol Biol. 2011:730:79-88.
- Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820-829.
- List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456-1465.
- Mallo M, Cervera J, Schanz J, et al. Impact of adjunct cytogenetic abnormalities for prognostic stratification in patients with myelodysplastic syndrome and deletion 5q. Leukemia. 2011;25:110-120.
- Coleman JF, Theil KS, Tubbs RR, et al. Diagnostic yield of bone marrow and peripheral blood FISH panel testing in clinically suspected myelodysplastic syndromes and/or acute myeloid leukemia: a prospective analysis of 433 cases. Am J Clin Pathol. 2011;135:915-920.
- De Smet D, Trullemans F, Jochmans K, et al. Diagnostic potential of CD34+ cell antigen expression in myelodysplastic syndromes. Am J Clin Path. 2012;138:732-743.
- Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes. Blood. 2012;119:5674-5677.
- Bacher U, Kern W, Alpermann T, et al. Prognosis of MDS subtypes RARS, RCMD and RCMD-RS are comparable but cytogenetics separates a subgroup with an inferior clinical course. Leuk Res. 2012;36:826-831.
- Malcovati L, Karimi M, Papaemmanuil E, et al. SFB31 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233-241.
- Papaemmanuil E, Cazzola M, Boultwood L, et al. Somatic SFB31 mutation in myelodysplasia with ringed sideroblasts. N Engl J Med. 2011;365:1384-1395.
- Alpermann T, Jeromin S, Haferlach C, et al. MDS and AML with ≥15% ring sideroblasts share overlapping features in cytogenetics but demonstrate different patterns and incidences of SF3B1 mutations. Blood. 2013;122:2776.
- Mihova D, Zhang L. Acute erythroid leukemia: a review. North American Journal of Medicine and Science. 2012;5:110-118.
- Park S, Picard F, Dreyfu F. Erythroleukemia: a need for a new definition. Leukemia. 2002;16:1399-1401.
- Cuneo A, Van Orshoven A, Michaux JL, et al. Morphologic, immunonologic and cytogenetic studies in erythroleukemia: evidence for multilineage involvement and identification of two distinct cytogenetic-clinicopathological types. Br J Haematol. 1990;75:346-354.
- Gangat N, Patnaik MM, Tefferi A. Myelodysplastic syndromes: contemporary review and how we treat. Am J Hematol. 2016;91:76-89.
- Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079-2088.
- Bejar R, Steensma DP. Recent developments in myelodysplastic syndromes. Blood. 2014;124:2793-2803.
- Greenberg P, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454-2465.
- Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25:3503-3510.
- Malcovati L, Della Porta MG, Strupp C, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica. 2011;96:1433-1440.
- Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113:1351-1361.
- Garcia-Manero G, Shah J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22:538-543.
- Quintas-Cardama A, Daver N, Kim H, et al. A prognostic model of therapy-related myelodysplastic syndrome for predicting survival and transformation to acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2014;14:401-410.
- Zeidan AM, Komrokji RS. There’s risk, and then there’s risk: the latest clinical prognostic risk stratification models in myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8:351-360.
- Garcia-Manero G. Myelodysplastic syndromes. 2015 update on diagnosis, risk-stratification and management. Am J Hematol. 2015;90:831-841.
- Papaemmanuil E, Gerstung M, Malcovati l, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616-3627.
- Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496-2506.
- Malcovati L, Della Porta MG, Pietra D, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114:3538-3545.
- Ali AM, Iverson N, Penson A, et al. Molecular genetic analysis of myelodysplastic syndromes (MDS) patients with ring sideroblasts (RS); independent confirmation of association of SF3B1 mutations with better prognosis. Blood. 2014;124:3237.
- Kulasekararaj AG, Smith AE, Mian SA, et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br J Haematol. 2013;160:660-672.
- Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705-2712.
- Patnaik MM, Lasho TL, Finke CM, et al. Splicesome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88:201-206.
- Naqvi K, Garcia-Manero G, Sardesai S, et al. Association of comorbidities with overall survival in myelodysplastic syndrome: development of a prognostic model. J Clin Oncol. 2011;29:2240-2246.
- Haferlach T, Nagata Y, Grossman V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241-247.
- Bejar R. Clinical and genetic predictors of prognosis in myelodysplastic syndromes. Haematologica. 2014;99:956-964.
- Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122:2943-2964.
- Wimazal F, Fonatsch C, Thalhammer R, et al. Idiopathic cytopenia of undetermined significance (ICUS) versus low-risk MDS: the diagnostic interface. Leuk Res. 2007;31:1461-1468.
- Kwok B, Hall JM, Witte JS, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126:2355-2361.
- Cargo C, Rowbotham N, Evans PA, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126:2362-2365.
- Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488-2498.
- Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472-1478.
- Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9-16.