 Progress in personalized medicine is currently taking place within a system of governmental regulation that was largely created before the term was even coined. Today’s regulatory framework, directed primarily by the Food and Drug Administration (FDA) and a handful of other federal and state agencies, was created incrementally over the course of the 20th century to meet various public health needs, from the thalidomide crisis of the 1960s to the HIV/AIDS crisis of the 1980s and 1990s.
Progress in personalized medicine is currently taking place within a system of governmental regulation that was largely created before the term was even coined. Today’s regulatory framework, directed primarily by the Food and Drug Administration (FDA) and a handful of other federal and state agencies, was created incrementally over the course of the 20th century to meet various public health needs, from the thalidomide crisis of the 1960s to the HIV/AIDS crisis of the 1980s and 1990s.
 As various regulatory gaps were filled over time, a complete system of regulation encompassing pharmaceuticals, medical devices, and diagnostic technologies emerged. This system, although comprehensive, was not designed by Congress with personalized medicine in mind, and thus it may be time to rethink how regulatory authorities are structured.
As various regulatory gaps were filled over time, a complete system of regulation encompassing pharmaceuticals, medical devices, and diagnostic technologies emerged. This system, although comprehensive, was not designed by Congress with personalized medicine in mind, and thus it may be time to rethink how regulatory authorities are structured.
 Here we provide a brief overview of the legislation that created the regulatory framework overseeing products in personalized medicine with the hope of improving understanding of why things are the way they are and how they might change to better align with the future needs of an advancing field.
Here we provide a brief overview of the legislation that created the regulatory framework overseeing products in personalized medicine with the hope of improving understanding of why things are the way they are and how they might change to better align with the future needs of an advancing field.
The Creation of the FDA and Drug Regulation
Pure Food and Drugs Act (1906)
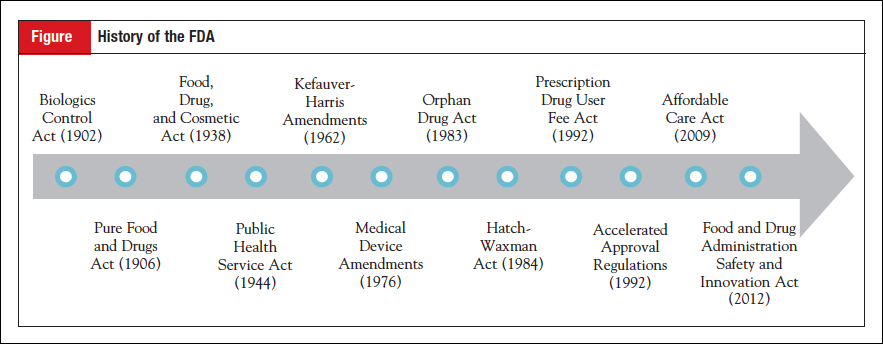
The law that created the nation’s first drug regulations was the 1906 Pure Food and Drugs Act, signed by Theodore Roosevelt after years of campaigning by progressives to address widespread medical fraud and food contamination. The leading advocate for reform was Harvey Washington Wiley, Chief Chemist in what is now the Department of Agriculture, who was among the first to champion the role of government in protecting the public from abuses in the market. One such abuse was the marketing of “patent medicines,” drugs that made lofty health claims but whose ingredients were withheld from doctors and patients. A series of articles in Collier’s magazine published in early 1906 exposed the ingredients of many of these “secret formula” medicines, showing that common remedies contained narcotics while others contained nothing but water and alcohol.
To address concerns about unknown ingredients in patent medicines, the 1906 Act introduced drug labeling requirements, but only for certain substances such as alcohol and opiates; all other ingredients were permitted to continue to be withheld from consumers. Additionally, the law prohibited “misbranding” of drugs, but the Supreme Court in United States v Johnson (1911) ruled that misbranding did not apply to false therapeutic claims, a decision that significantly diminished the impact of the legislation, as assertions that drugs were cure-alls went uncontested. Moreover, it would not be until the 1960s that false therapeutic claims were effectively curtailed by the FDA. The 1906 Act was primarily about policing fraud, not assuring drug safety; nothing in the law could prevent harmful drugs from entering the market.
Food, Drug, and Cosmetic Act (1938)
Significant action to overhaul the 1906 Act did not begin until 1933, when a bill was drafted that would extend misbranding provisions to advertisements, require labels to display all ingredients, not just addictive ones, and, most importantly, require drugmakers to submit evidence that their products were safe before selling them. FDA officials made the case for increased regulation with an exhibit that came to be known in the press as the “Chamber of Horrors,” a collection of the most egregious safety issues associated with drugs that highlighted dangers that were currently beyond the reach of the law. Although these efforts drew attention to reform, a public health crisis was the primary impetus for passage of new regulations. In 1937 the antibiotic sulfanilamide, having been combined with the solvent diethylene glycol, killed over 100 people, many of them children. Congress, seeking to prevent future tragedies, passed the Food, Drug, and Cosmetic Act, which was signed by Franklin Roosevelt on June 15, 1938.
The 1938 Act established premarket review of safety for new drugs, changing the FDA’s position from responding to harm to attempting to prevent it.1 It also led to the creation of a scientifically minded pharmaceutical industry, given its requirement that drug makers produce evidence about the effects of their products.2 However, like its predecessor, the 1938 Act had flaws that would need to be addressed by future policymakers. The first was that only safety, not both safety and effectiveness, was required to be demonstrated. The closest it came was to tweak misbranding language from the 1906 Act to include false therapeutic claims, but these were dealt with in the courts, an inappropriate forum to assess the merits of a drug. The second flaw was that applications for approval became effective automatically after 60 days, leaving the FDA only 2 months to decide if a drug was safe.1
Kefauver-Harris Amendments (1962)
FDA officials, well aware of the limitations of the 1938 Act, began to lobby members of Congress and draft legislation in the late 1950s to address gaps in oversight.3 These efforts coincided with a series of hearings on pharmaceutical monopolies and price fixing led by Senator Estes Kefauver of Tennessee. A number of proposals emerged from this spike in attention on the FDA, but, much like the 1938 Act, congressional action only took place in the wake of public outcry. A front-page article in the Washington Post in the summer of 1962 told the story of how an FDA official, named Frances Kelsey, refused to give a positive opinion on a drug called thalidomide, an act that came to be viewed as heroic after the drug, often used to treat morning sickness, was found to have caused hundreds of birth defects in children in Western Europe. The story reminded the public of the importance of drug safety laws, while also lifting the reputation of the FDA as a protector of public health, embodied in the maternal persona of Francis Kelsey.3
The 1962 Kefauver-Harris Amendments to the 1938 Act, passed shortly after the thalidomide incident, made 2 major changes to drug regulation. First, they overturned the automatic approval provision of the 1938 Act, revising the existing premarket notification system into a premarket approval system in which the FDA now held veto power over new drugs entering the market.1 This provision inaugurated FDA’s gatekeeping power, requiring all new drugs to pass through the FDA on the way to market. Second, drugs now had to demonstrate evidence of effectiveness as well as safety, dramatically increasing the amount of time, resources, and scientific expertise required to develop a new drug.
Birth of the Modern Clinical Trial System
To be implemented, the 1962 Amendments required interpretation of the legislative text, which stated effectiveness had to be derived from “substantial evidence” in “adequate and well-controlled investigations.” Drug-makers looked to the FDA to lay the ground rules for how they should conduct their experiments, and as a result, the FDA’s interpretation of concepts like “efficacy” played a central role in shaping how clinical trials would be conducted moving forward. The concept of 3 phases of experiment emerged in the wake of the new law and was adopted by the FDA, becoming the default method for studying medicine in humans ever since.
Filling Regulatory Gaps: Biologics, Devices, and Diagnostics
Slightly over 20% of consumer spending in the United States is on products regulated by the FDA. Past Congresses have given the FDA authority to regulate a spectrum of other medical products beyond food and drugs, from biologics to in vitro diagnostics. However, the creation of today’s regulatory framework took place slowly over the course of the 20th century, with separate categories of products coming under government oversight incrementally as technology advanced. Periodic adjustment to the FDA’s governing statute continues to occur as science evolves and new types of products come on the market.
Biologics
The first regulations concerning biologics actually preceded the 1906 drug law by 4 years; in 1902 the Biologics Control Act required purveyors of vaccines to be licensed and gave the Hygienic Laboratory-renamed the National Institutes of Health (NIH) in 1930-authority to establish standards for the production of vaccines. Regulation of vaccines and other biologic products would be housed in the NIH until 1972, when it was transferred to the FDA. In 1944, the Public Health Service Act expanded regulation of biologics to the products themselves, not just the bodies that manufactured them, but standards for effectiveness equivalent to those for drugs were not imposed until the move to the FDA in 1972. Biologics are currently overseen by the Center for Biologics Evaluation and Research at the FDA, which exists alongside parallel centers for drugs and devices. An internal reorganization of the FDA in 2004 resulted in the transfer of regulation of some therapeutic biologics, including monoclonal antibodies, to the Center for Drug Evaluation and Research, allowing for the streamlining of oversight of many cancer agents.
Devices
Medical devices first came under government regulation in the 1938 Food, Drug, and Cosmetic Act, although, as the law’s name reveals, they were not yet considered a separate category of product, defined instead under the term “drug.” The 1938 Act provided the FDA with authority to take legal action against the adulteration and misbranding of medical devices, although it did not contain a premarket notification provision for devices, as it did for drugs.4 When the 1962 Kefauver-Harris Amendments were passed, there were rumors that Congress would consider a companion bill requiring premarket approval for medical devices shortly thereafter. However, it took 15 years for comprehensive legislation to be passed. The 1976 Medical Device Amendments created an alternative regulatory approach that involved classifying devices according to risk and strengthened the provisions of the 1938 Act to include premarket review of those devices that fell into the high-risk category.
Diagnostics
In implementing the 1976 Medical Device Amendments, the FDA was required to conduct an inventory and classification of all existing devices to fit products into risk categories that would then inform whether a device needed to undergo the premarket review process. The FDA classified in vitro diagnostics (IVDs) as medical devices, and many IVDs that have become central to personalized medicine, such as pharmacogenomic tests, fall into FDA’s highest risk category. A separate category of tests, called laboratory developed tests (LDTs), were not initially regulated by FDA but rather the Centers for Medicare & Medicaid Services acting under the Clinical Laboratory Improvement Amendments of 1988. The FDA has claimed jurisdiction over all tests, both IVDs and LDTs, but has exercised enforcement discretion with regard to the latter until very recently, when it proposed extending oversight to LDTs.5 As laboratory medicine has increased in complexity, a greater number of LDTs are being considered high-risk tests due to their role in diagnosing disease and steering treatment decisions.
Spurring Innovation and Patient Access
Long before the “10 years, 1 billion dollars” figure was attached to drug development, there was a general view that new drugs appeared rather slowly and patients suffered as a result, especially those with deadly diseases. In the 1980s and 1990s, upon the urgings of patient groups and observers who felt more could be done to bring drugs to patients quickly, policymakers passed a series of bills and administrative reforms that promoted patient access to new drugs.
Drugs for Rare Diseases
One of the first pieces of legislation to promote innovation in the pharmaceutical industry was the Orphan Drug Act of 1983, passed in response to concerns that companies lacked incentives to develop drugs with limited commercial value. Primarily intended for rare diseases, the law has since been applied to many development programs for biomarker-enriched cancer populations, such as EGFR- and ALK-positive lung cancer. Under the Orphan Drug Act, Congress defines a rare disease or condition as affecting fewer than 200,000 people in the United States or for which there is no reasonable expectation that the sales of the drug treatment will recover the costs.6 Drugs that are designated as orphan products benefit from 2 years’ additional marketing exclusivity (7 years vs the standard 5 years), federal grants to conduct clinical trials, and tax credits for clinical development costs. The orphan designation has been granted widely in the field of oncology, with one report finding that 27% of all orphan approvals between 1983 and 2009 were for cancer drugs.7
Generics and Biosimilars
The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act for Senator Orrin Hatch and Representative Henry Waxman, gave rise to the modern generic drug market. It was designed with 2 purposes in mind: 1) to preserve incentives to develop new drugs, and 2) to make low-cost generics widely available. The Act offset an unintended consequence of the 1962 Kefauver-Harris Amendments that greatly increased clinical development time, which in turn shortened the remaining patent life of medicines once they entered the market. The Hatch-Waxman Act “restored” some of the lost patent life, thereby increasing financial incentives to develop new drugs. In addition, the Act made it possible for manufacturers of generic products to apply for approval without demonstrating safety and effectiveness, requiring only that generics are shown to be the “same” as and bioequivalent to brand name products.8
These dual aims of enhancing innovation and expanding patient access were also reflected in the Biologics Price Competition and Innovation Act of 2009, which Congress created to promote competition in the biologics market once products go off patent. The complexity of biologic products prevents them from being replicated in the same fashion as small molecule drugs, so instead of demonstrating bioequivalence, the law requires evidence of “biosimilarity,” defined as the absence of clinically meaningful differences between the biosimilar and the reference product. The first biosimilar approval in the United States was in March 2015, and a number of other products are currently in development, although many developers are anticipating further guidance from the FDA on how to best demonstrate biosimilarity.
Speeding Review and Development Times
Major changes to drug policy took place in response to the HIV/AIDS crisis of the 1980s and 1990s. The accelerated approval regulations, instituted in 1992, made it possible for drugs intended to treat serious or life-threatening diseases to be approved more quickly on the basis of surrogate end points. Drugs that receive accelerated approval must show evidence of improvement over available therapy based on a surrogate end point that is reasonably likely to predict clinical benefit.9 These regulations, which changed approval standards, were initially brought about by administrative rulemaking rather than legislation-accelerated approval was not codified in statute until 2012. Accelerated approval has been used most widely in the field of oncology, with one-third of all oncology approvals between 2002 and 2012 approved via the accelerated pathway.10 Oncology has benefited most from this program largely due to the identification of numerous surrogate end points that can reasonably predict survival, such as progression-free survival and response rate.
Also taking place in 1992 was passage of the Prescription Drug User Fee Act (PDUFA), which has lent consistency and predictability to drug review times. Twenty years earlier in 1971, critics of the FDA coined the term “drug lag” to describe instances in which new medicines were made available in Europe prior to the United States. The drug lag became a perennial talking point among critics of the agency as evidence of regulation impeding patient access. In 1980, a report published by the General Accounting Office disputed this narrative, attributing backlogged new drug applications to inadequate resources. Rather than increase direct appropriations to the FDA, policymakers settled on a “user fee” program, wherein the pharmaceutical industry would provide funds to hire additional FDA reviewers in return for assurances of timely reviews of new drug applications. PDUFA had an immediate impact, speeding up review times and allowing the FDA to consistently meet its 10-month goal for standard reviews and 6-month goal for priority applications.11 Due to the program’s success, additional user fee programs have been established for generic drugs, medical devices, and biologics. The law has a sunset clause, requiring it to be reauthorized every 5 years to allow user fees to be renegotiated based on the FDA’s performance in meeting review timelines. Each PDUFA reauthorization (there have been 5 so far) has presented an opportunity to pass additional legislation related to the FDA, and, in recent years, such add-ons have focused on promoting innovation in the pharmaceutical industry.
The most recent reauthorization of PDUFA took place in 2012 and was accompanied by a series of reforms to the FDA intended to spur innovation and speed drug development. The authorizing law, called the Food and Drug Administration Safety and Innovation Act (FDASIA), created a new method, called the breakthrough therapy designation, for the FDA to speed the development of certain drugs. To receive the designation, a drug must be intended for a serious or life-threatening disease and early clinical evidence (usually from phase 1 or 2 trials) indicates that the drug may provide a substantial improvement over available therapy. Designed as a way for the FDA to expedite the development of drugs that have the potential to be transformative, the breakthrough therapy designation confers increased communication with high-level FDA officials who can provide advice on development programs and the most efficient path forward. The designation has been granted to over 100 drug development programs, and over 30 have been approved, with more than one-third of approvals for anticancer agents.
Also included in FDASIA was a provision that created the patient-focused drug development initiative at FDA, which brought patients together in disease-specific meetings to share their experiences with FDA officials. The goal of the initiative is to use this “patient experience data” to inform clinical trial design, end points, and risk-benefit calculations to better reflect patient needs. Two oncology-specific meetings have already been held for lung and breast cancer patients, and another is planned for neuropathic pain associated with peripheral neuropathy in 2016.
Looking Forward
The current regulatory framework, although comprehensive, came about in a piecemeal fashion through a patchwork of laws granting the FDA authority to regulate various new types of medical products. As a consequence, the agency’s structure is oriented around the products it regulates and is divided into multiple centers, each devoted to oversight of a different product. While this structure has allowed for an aggregation of product-related expertise, it does not fully reflect the current multimodal approach to medical care. In the field of oncology, for example, therapeutics are being developed using genetic information with increased frequency, a trend that involves the concurrent use of drugs and molecular diagnostics. In its current form, the FDA is not optimally positioned to address the coordinated use of a spectrum of technologies and interventions common in medical practice today.
Thus, rather than maintaining a product-oriented approach to regulating new treatments, the FDA should adopt a patient-centered orientation to reflect the current multimodal approach to patient care. This should include an organizational realignment at the FDA based on major disease areas. Housing functions and expertise according to disease areas would better reflect how products are used in practice and would enhance collaborative interactions and streamline administrative processes. Such a patient-oriented realignment will also allow for enhanced interactions with patients and the external biomedical community who already approach disease states holistically rather than by product type. Increased staffing and resources that go beyond the review functions should be provided to support this type of realignment at the agency to ensure optimal implementation and long-term success.
Conclusion
Over the course of the 20th century and into the 21st, a system of regulation was established for a broad spectrum of medical products. Although crises typically preceded passage of new laws, advancing science and the development of new technologies were what shaped the content of reform efforts. In some cases, changing science enabled policymakers to explore ways of making the development process more efficient, as was the case for the accelerated approval regulations, which stemmed from an understanding of surrogate end points, and the breakthrough therapy designation, which was inspired by dramatic improvements seen in early-phase trials. In other cases, policies clearly shaped the subsequent conduct of science, such as the Kefauver-Harris Amendments, which inaugurated the concept of phased drug development, and the Orphan Drug Act, which stimulated the development of tools to evaluate drug efficacy in small populations.
Recent reform efforts have similarly focused on ways to promote scientific advances with legislation. As noted above, each reauthorization of the PDUFA has enabled lawmakers to consider legislation related to medical product regulation. Members of the House Committee on Energy and Commerce and the Senate Committee on Health, Education, Labor and Pensions are currently weighing a host of proposals that may be coupled with the 6th PDUFA. This will present a new opportunity to assess the current regulatory framework, and if Congress determines it necessary, to make adjustments.
References
- Merrill RA. The architecture of government regulation of medical products. Virginia Law Rev. 1996;82:1753-1866.
- Hilts PJ. Protecting America’s Health: The FDA, Business, and One Hundred Years of Regulation. New York, NY: Knopf; 2003.
- Carpenter D. Reputation and Power: Organizational Image and Pharmaceutical Regulation at the FDA. Princeton, NJ: Princeton University Press; 2010.
- Hutt PB. A History of Government Regulation of Adulteration and Misbranding of Medical Devices. In: Estrin NF, ed. The Medical Device Industry: Science, Technology, and Regulation in a Competitive Environment. New York, NY: Marcel Dekker Inc; 1990.
- US Food and Drug Administration. Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs). 2014.
- Field MJ, Boat TF, eds. Rare Diseases and Orphan Products: Accelerating Research and Development. Washington, DC: Institute of Medicine; 2010.
- Wellman-Labadie O, Zhou Y. The US Orphan Drug Act: rare disease research stimulator or commercial opportunity? Health Policy. 2010;95:216-228.
- Danzis SD. The Hatch-Waxman Act: history, structure, and legacy. Antitrust Law Journal. 2003;71:585-608.
- Dagher R, Johnson J, Williams G, et al. Accelerated approval of oncology products: a decade of experience. J Natl Cancer Inst. 2004;96:1500-1509.
- Shea MB, Roberts SA, Walrath JC, et al. Use of multiple endpoints and approval paths depicts a decade of FDA oncology drug approvals. Clin Cancer Res. 2013;19:3722-3731.
- US Food and Drug Administration. FY 2014 PDUFA Performance Report. www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceRe ports/ucm440180.htm. Accessed February 17, 2016.